
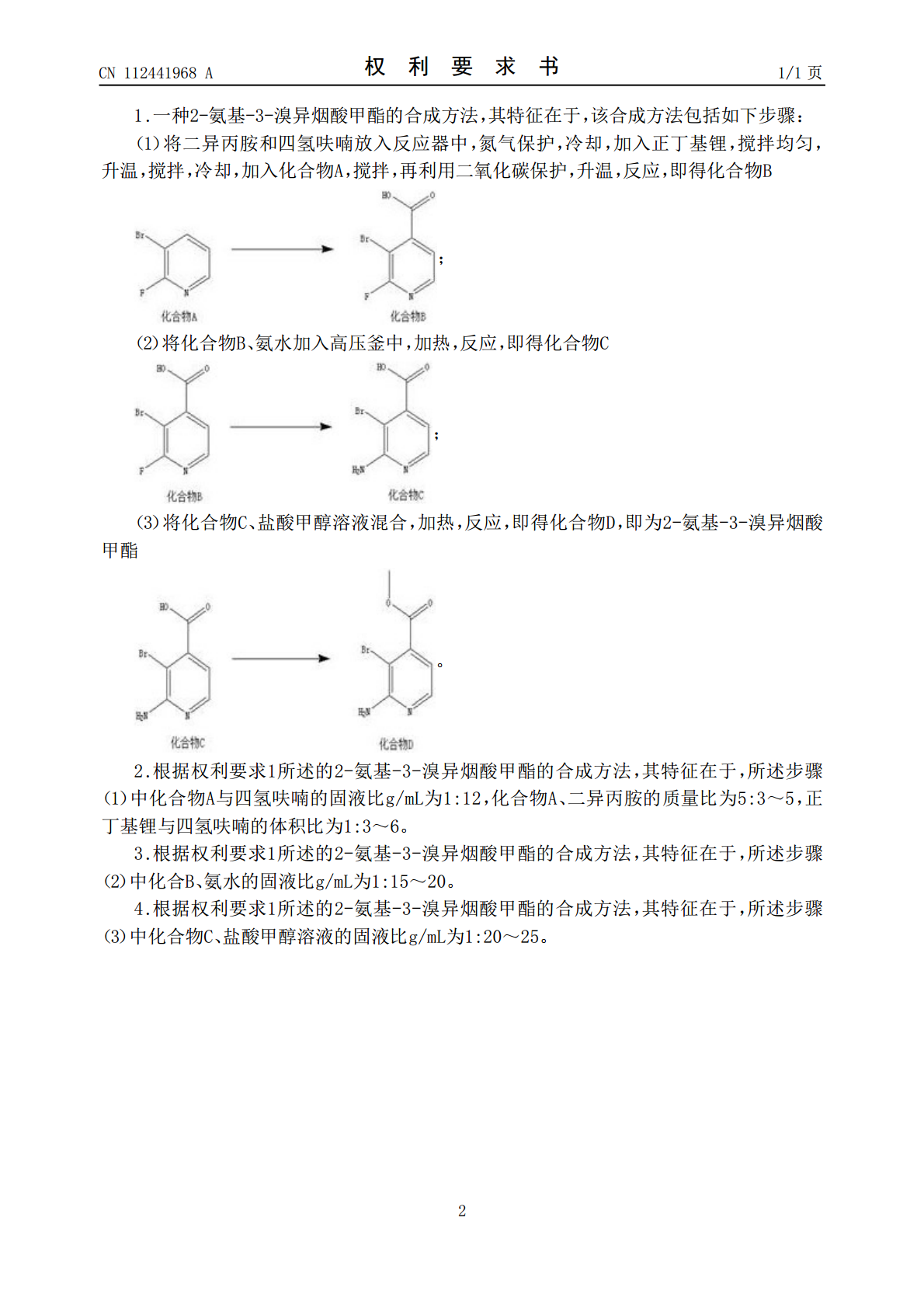
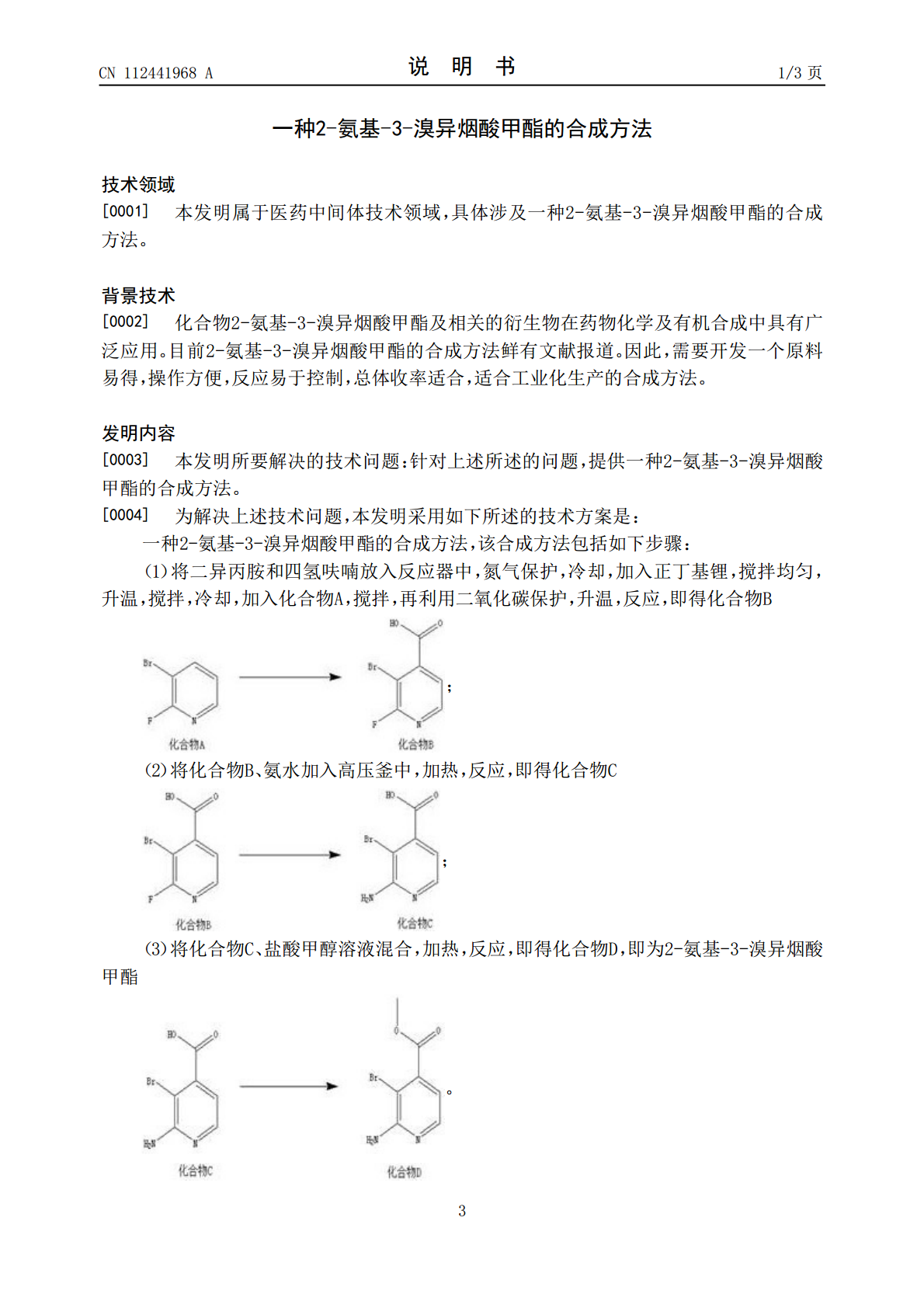
一种2-氨基-3-溴异烟酸甲酯的合成方法.pdf

雨星****萌娃


1/5
2/5
3/5

4/5

5/5
在线预览结束,喜欢就下载吧,查找使用更方便
相关资料

一种2-氨基-3-溴异烟酸甲酯的合成方法.pdf
本发明属于医药中间体技术领域,具体涉及一种2‑氨基‑3‑溴异烟酸甲酯的合成方法。本发明以化合物A作为起始原料,通过与二异丙胺等进行反应,形成化合物B,再通过化合物B的反应等后续反应,形成了2‑氨基‑3‑溴异烟酸甲酯,本发明首次提出了2‑氨基‑3‑溴异烟酸甲酯的合成方法;本发明所得到的产物收率较高,纯度好。
一种3-氨基异烟酸甲酯的合成方法.pdf
本发明公开了一种3‑氨基异烟酸甲酯的合成方法,以4‑三氟甲基烟酸为起始原料,依次经过酰化缩合串联、Hofmann降解、水解、酯化反应得到所述3‑氨基异烟酸甲酯。本发明合成方法反应条件温和、收率高、成本低、原料易得、可工业化生产,且氯化亚砜反应液可以反复套用,提高原料的利用率,减少资源浪费,减少污染,最大限度地降低整体工艺的生产成本,具有极高的应用价值。

一种快速高效合成3-氨基异烟酸甲酯的方法.pdf
本发明属于化学制药领域,具体公开一种快速高效合成3‑氨基异烟酸甲酯的方法,经硝化、还原、酯化得到所述3‑氨基异烟酸甲酯。本发明所述快速高效合成3‑氨基异烟酸甲酯的方法,反应条件温和,总体反应速率高,总收率高,催化剂中主成分钯碳可以反复利用,提高原料的利用率,减少资源浪费,最大限度地降低整体工艺的成产成本,具有极高的应用。

一种3-氨基异烟酸甲酯的高收率合成方法.pdf
本发明属于化学制药领域,具体公开一种3‑氨基异烟酸甲酯的高收率合成方法,以4‑吡啶羧酸为原料,经溴化、氨化、酯化得到所述3‑氨基异烟酸甲酯。本发明所述3‑氨基异烟酸甲酯的高收率合成方法,反应条件温和,总收率高,3‑溴‑4‑吡啶羧酸反应废弃滤液可以反复套用,提高原料的利用率,减少资源浪费,最大限度地降低整体工艺的成产成本,具有极高的应用。

一种4-氨基烟酸甲酯的高收率合成方法.pdf
本发明公开了一种4‑氨基烟酸甲酯的高收率合成方法,以3,4‑吡啶二羧酸为原料,经过分子内脱水取代、氨气氨化、改良型NBS霍夫曼重排后水解得到所述4‑氨基烟酸甲酯。本发明合成方法,操作更加简单,反应条件温和,总收率也更高,具有极高的应用价值。