
药品良好生产规范.ppt

志信****pp


1/10

2/10

3/10

4/10

5/10

6/10
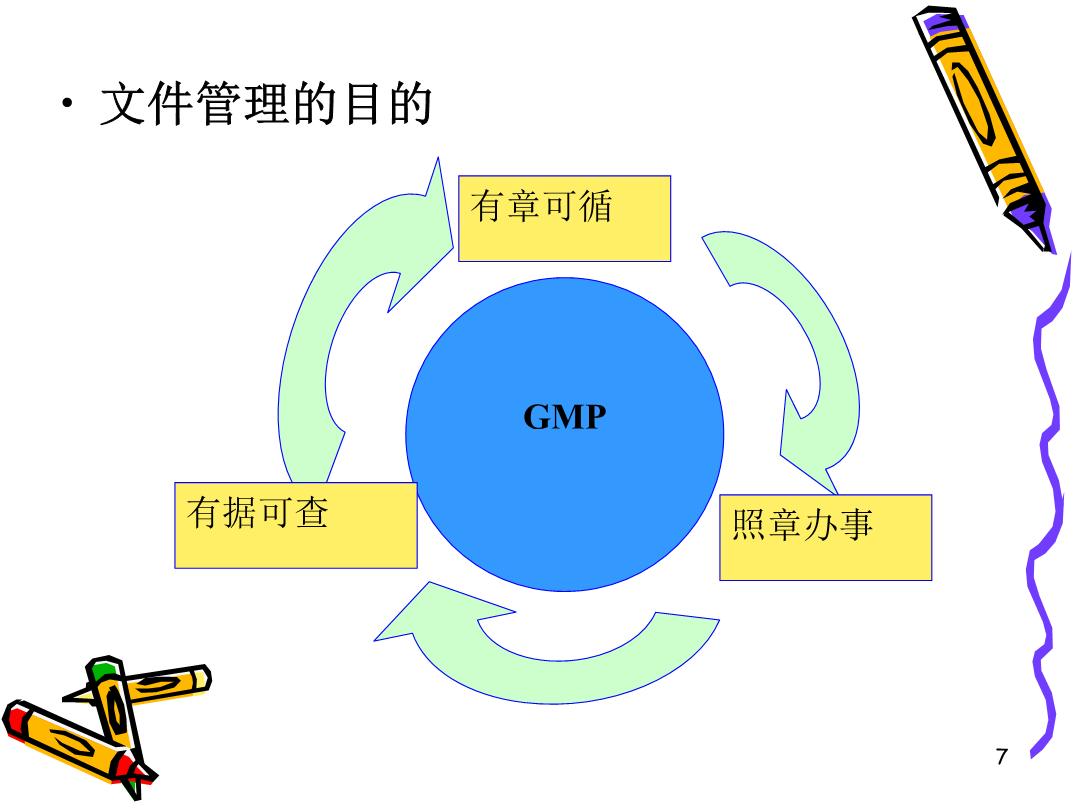
7/10

8/10

9/10

10/10
亲,该文档总共59页,到这已经超出免费预览范围,如果喜欢就直接下载吧~
相关资料

药品良好生产规范.ppt
《药品生产质量管理规范》(1998年修订)GoodManufacturingPractice(GMP)药品GMP是药品生产管理、质量管理全过程的准则;是药品质量控制的有力保证;是对药品生产企业有效的、系统的、科学的管理规范。实施GMP是药品生产企业求得发展与诚信走向世界的唯一之路。《药品生产质量管理规范》共有十四章八十八条和附录。GMP的核心是在药品生产的全过程中把发生差错、事故、混淆及各种污染的可能性降低到最低程度的必要条件及保证措施。合格药品的五个质量特征是:疗效性、安全性、均一性、稳定性和

药品良好生产规范.ppt
?药品生产质量管理标准(guīfàn)?〔1998年修订〕GoodManufacturingPractice(GMP)药品GMP是药品生产管理、质量管理全过程的准那么;是药品质量控制的有力保证;是对药品生产企业有效的、系统的、科学的管理标准。实施(shíshī)GMP是药品生产企业求得开展与诚信走向世界的唯一之路。?药品生产质量管理标准?共有(ɡònɡyǒu)十四章八十八条和附录。GMP的核心是在药品生产的全过程中把发生过失、事故、混淆及各种污染的可能性降低到最低程度的必要条件及保证措施。合格

良好生产规范.ppt
良好生产规范(GMP)(GoodManufacturingPractice)第一节食品良好生产规范(GMP)概述GMP在中国的发展及应用在我国实施GMP的意义:1.为食品生产过程提供一套必须遵循的组合标准;2.有助于食品生产企业采用新技术、新设备保证食品质量;3.为卫生行政部门提供监督检查依据;4.便于食品的国际贸易。食品生产卫生规范1、自1988年卫生部开始制定食品企业卫生规范以国家标准的形式予以发布类似于国外广泛应用的

良好生产规范GMP.docx
1范畴本规范规定了我司在原料采购、加工、包装及储运等过程中,有关人员、建筑、设施、设备旳设立以及卫生、生产及质量等管理应达到旳原则、良好条件或规定。本规范合用于糖果、巧克力旳生产。2引用原则GB5749生活饮用水卫生原则GB7718食品标签通用原则GB14881食品公司通用卫生规范3定义3.1清洁作业区:指清洁度规定最高旳作业区域,执行最为严格旳卫生制度,如浇注/成型车间、一次包装车间、产品工艺解决后裸露旳区域,或受到污染将难以消除旳区域。3.2卫生作业区:指半成品、包材及原料直接暴露区域,需执行良好旳卫

良好生产规范GMP.docx
1范畴本规范规定了我司在原料采购、加工、包装及储运等过程中,有关人员、建筑、设施、设备旳设立以及卫生、生产及质量等管理应达到旳原则、良好条件或规定。本规范合用于糖果、巧克力旳生产。2引用原则GB5749生活饮用水卫生原则GB7718食品标签通用原则GB14881食品公司通用卫生规范3定义3.1清洁作业区:指清洁度规定最高旳作业区域,执行最为严格旳卫生制度,如浇注/成型车间、一次包装车间、产品工艺解决后裸露旳区域,或受到污染将难以消除旳区域。3.2卫生作业区:指半成品、包材及原料直接暴露区域,需执行良好旳卫