
一种制备2,4‑二氨基‑6‑[2‑(2‑甲基‑1‑咪唑)乙基]‑1,3,5‑三嗪的方法.pdf

涵蓄****09


1/4

2/4

3/4

4/4
在线预览结束,喜欢就下载吧,查找使用更方便
相关资料

一种制备2,4‑二氨基‑6‑[2‑(2‑甲基‑1‑咪唑)乙基]‑1,3,5‑三嗪的方法.pdf
本发明公开了一种制备2,4‑二氨基‑6‑[2‑(2‑甲基‑1‑咪唑)乙基]‑1,3,5‑三嗪的方法,它以2‑甲基咪唑为原料,经丙烯腈加成得到1‑氰乙基‑2‑甲基咪唑,1‑氰乙基咪唑再与双氰胺环合经两步反应合成得到目标产物。与现有技术相比,本发明采用2‑甲基咪唑原料,经加成、环合反应合成目标产物,该工艺收率高、操作简便等优点,易于工业化大生产。

6-甲基-1,3,5-三嗪-2,4-二胺及其制备方法.pdf
本发明提供了一种6‑甲基‑1,3,5‑三嗪‑2,4‑二胺及其制备方法,其制备方法包括:将双氰胺、乙腈、碱和有机溶剂依次加入容器中混合,搅拌下反应,反应液冷却结晶过滤,潮品经二次结晶后过滤干燥即可;本发明通过更换溶剂或溶剂混合体系,可以降低反应温度,从而避免高温,尤其是高压反应的条件;选用的溶剂体系可改善反应体系的粘稠程度,且工艺条件的稳定性对产品质量控制提供了保障;本发明的6‑甲基‑1,3,5‑三嗪‑2,4‑二胺的制备方法中以双氰胺、乙腈为原料,在碱催化作用下加热合环,经后处理得到质量合格的产品。原料廉价

一种制备2-乙基-4-甲基咪唑的方法.pdf
本发明公开了一种制备2-乙基-4-甲基咪唑的方法,以亚铜盐为催化剂,2-氨基丙醛缩二醇与丙腈在50~150℃下搅拌反应2~15小时,再向反应体系中加入醇类溶剂和浓盐酸,在40~80℃下继续搅拌反应1~6小时,即制得2-乙基-4-甲基咪唑。本发明方法采用一锅法,原料成本低、操作简便、反应条件温和、污染少、收率高等优点,易于工业化生产。

3-氨基-2,4-二甲基噻吩的制备方法.pdf
本发明涉及氯乙酰胺制备领域,公开了一种3‑氨基‑2,4‑二甲基噻吩的制备方法,该方法包括以下步骤,1)在酸酐和碱的存在下,将2,5‑二甲基‑3‑噻己二酸进行环合脱羧得到2,4‑二甲基四氢噻吩‑3‑酮;2)将2,4‑二甲基四氢噻吩‑3‑酮与羟胺或其盐缩合后重排得到3‑氨基‑2,4‑二甲基噻吩。通过本发明的方法,能够提供一种收率高,容易大规模生产的适合工业化的3‑氨基‑2,4‑二甲基噻吩的制备方法。
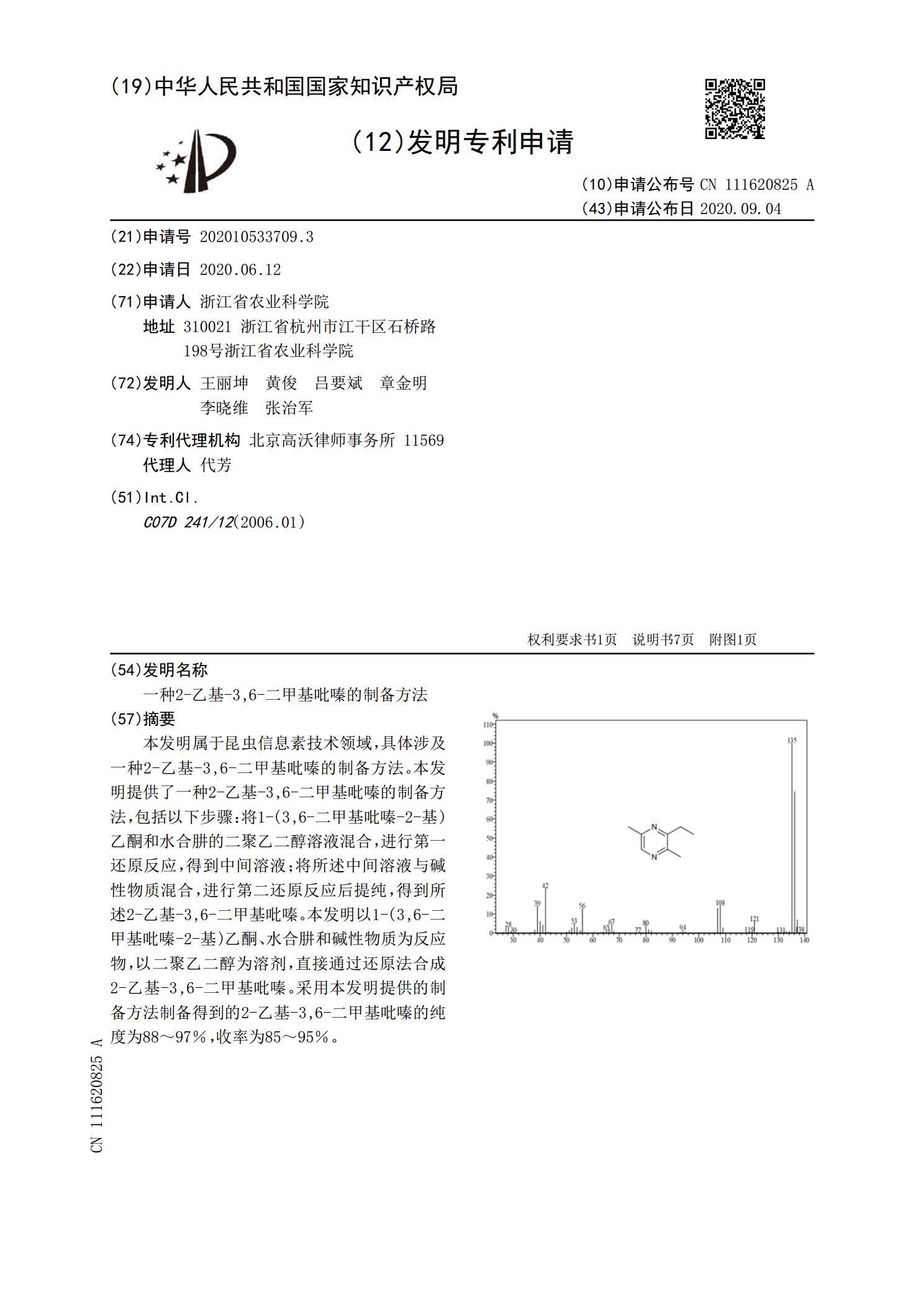
一种2-乙基-3,6-二甲基吡嗪的制备方法.pdf
本发明属于昆虫信息素技术领域,具体涉及一种2‑乙基‑3,6‑二甲基吡嗪的制备方法。本发明提供了一种2‑乙基‑3,6‑二甲基吡嗪的制备方法,包括以下步骤:将1‑(3,6‑二甲基吡嗪‑2‑基)乙酮和水合肼的二聚乙二醇溶液混合,进行第一还原反应,得到中间溶液;将所述中间溶液与碱性物质混合,进行第二还原反应后提纯,得到所述2‑乙基‑3,6‑二甲基吡嗪。本发明以1‑(3,6‑二甲基吡嗪‑2‑基)乙酮、水合肼和碱性物质为反应物,以二聚乙二醇为溶剂,直接通过还原法合成2‑乙基‑3,6‑二甲基吡嗪。采用本发明提供的制备方