
线粒体脑肌病.ppt

15****92


1/10

2/10

3/10

4/10

5/10

6/10

7/10

8/10

9/10
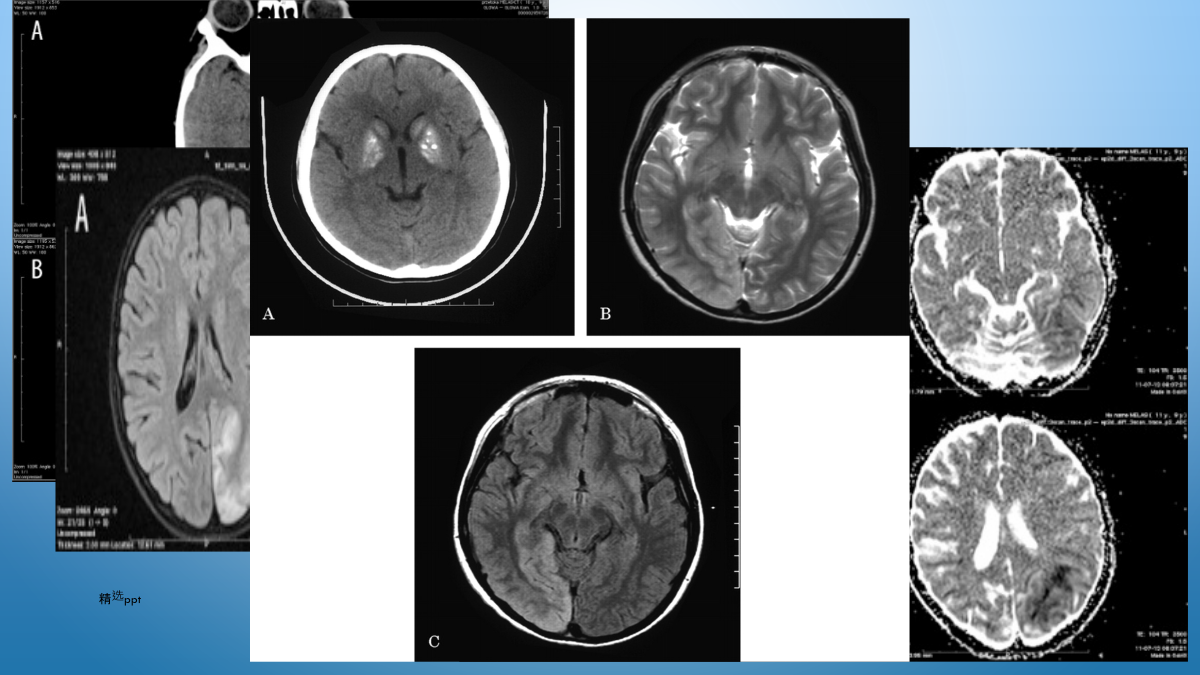
10/10
亲,该文档总共20页,到这已经超出免费预览范围,如果喜欢就直接下载吧~
相关资料

线粒体脑肌病.ppt
线粒体脑肌病病例特点:病例特点:影像资料:影像资料:定位诊断:定性诊断:诊断:线粒体脑肌病(MitochondrialEncephalopathy)线粒体简介:病因:临床表现及分型:下列各型症状可互相重叠诊断依据:鉴别诊断1鉴别诊断2鉴别诊断3线粒体脑肌病的治疗资料可以编辑修改使用学习愉快!课件仅供参考哦,实际情况要实际分析哈!感谢您的观看

线粒体脑肌病.ppt
线粒体(脑)肌病mitochondrialencephalomypathy一、概述二、发病机制三、临床表现2、慢性进行性眼外肌瘫痪(CPEO):多种遗传方式,儿童起病,以眼睑下垂为首发症状,缓慢进展为眼外肌瘫痪,眼球运动障碍乃至完全固定,部分患者可有咽部肌肉和四肢无力。3、Kearns-sayre综合征(kss):三联征:20岁以前发病,CPEO、视网膜色素变性。具备如上三种再加上以下之一即可诊断:心脏传导阻滞、小脑症状、脑脊液蛋白大于100mg/dl。常伴有神经性耳聋、智能减退等。4、肌阵挛性癫痫伴肌肉

线粒体脑肌病.ppt
线粒体脑肌病概念病因病机生理病理临床表现辅助检查影像学表现影像学表现神经电生理病理活检线粒体基因检测诊断鉴别诊断治疗其他治疗药物治疗运动疗法--括阻力和耐力训练。L-精氨酸预后伴发症鉴别诊断诊断

线粒体脑肌病.ppt
线粒体脑肌病概念病因病机生理病理临床表现辅助检查影像学表现影像学表现神经电生理病理活检线粒体基因检测诊断鉴别诊断治疗其他治疗药物治疗运动疗法--括阻力和耐力训练。L-精氨酸预后伴发症鉴别诊断诊断

线粒体脑肌病.ppt
线粒体脑肌病一定义60年代,Luft等于1962年首次报道了线粒体肌病由线粒体氧化磷酸化偶联障碍引起的肌病和非甲状腺功能亢进性高代谢状态随后,En-gel在光镜下观察不整边红纤维(sRRF)1977年,Shapria首次提出了线粒体脑肌病的诊断80年代,Wallace在Leber遗传性视神经病中证实了线粒体DNA(mitochondrialDNAmtD-NA)的点突变Holt在线粒体脑肌病中证实有mtDNA的减失从而(cóngér)使人们对线粒体病有了崭新的认识。二线粒体线粒体的形态(xíngtài)线粒