
P型掺杂BaSnO_3的电子结构和光学性能的第一性原理研究.pdf

qw****27


1/5

2/5
3/5
4/5

5/5
在线预览结束,喜欢就下载吧,查找使用更方便
相关资料

P型掺杂BaSnO_3的电子结构和光学性能的第一性原理研究.docx
P型掺杂BaSnO_3的电子结构和光学性能的第一性原理研究摘要:本文基于第一性原理研究了P型掺杂BaSnO3的电子结构和光学性能。通过使用密度泛函理论计算了掺杂前后的材料的电子结构及其能带结构。结果表明,掺杂P元素能够显著影响材料的电子结构,从而使其变成P型导体。此外,我们研究了掺杂后材料的吸收、透射和反射特性,发现其在可见光和近红外光波段的吸收率显著增强。这些发现表明,P型掺杂BaSnO3具有潜在的应用价值,尤其是在太阳能电池等领域。关键词:第一性原理、掺杂、BaSnO3、电子结构、光学性能引言:BaS

P型掺杂BaSnO_3的电子结构和光学性能的第一性原理研究.pdf
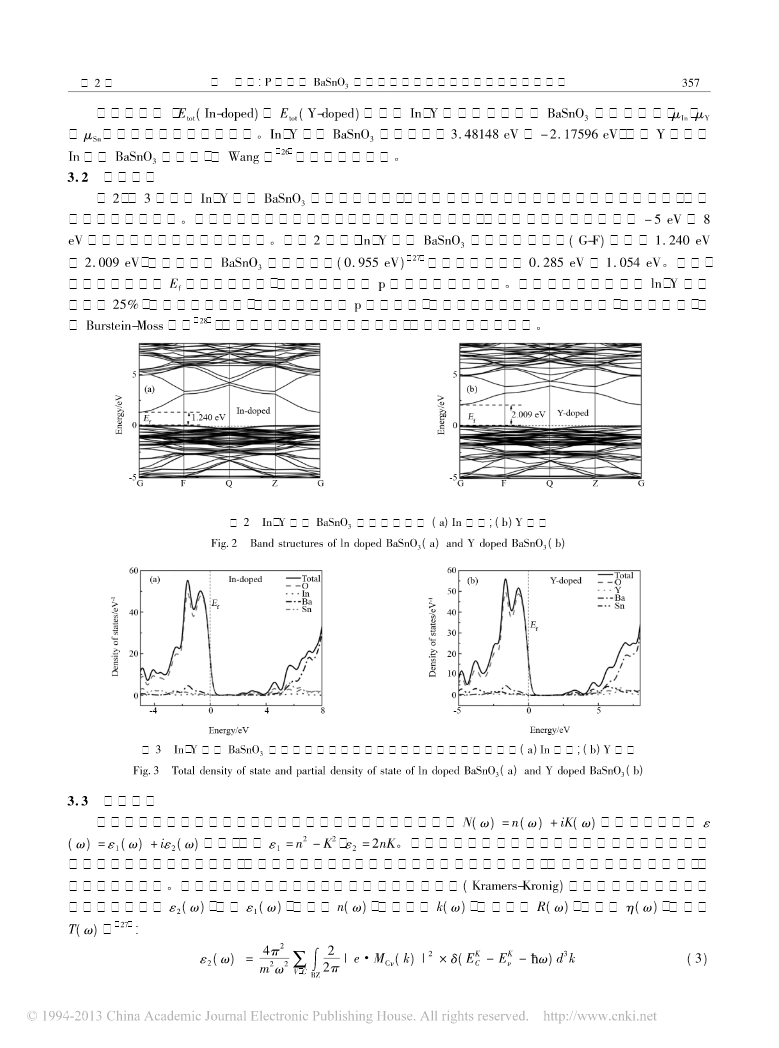
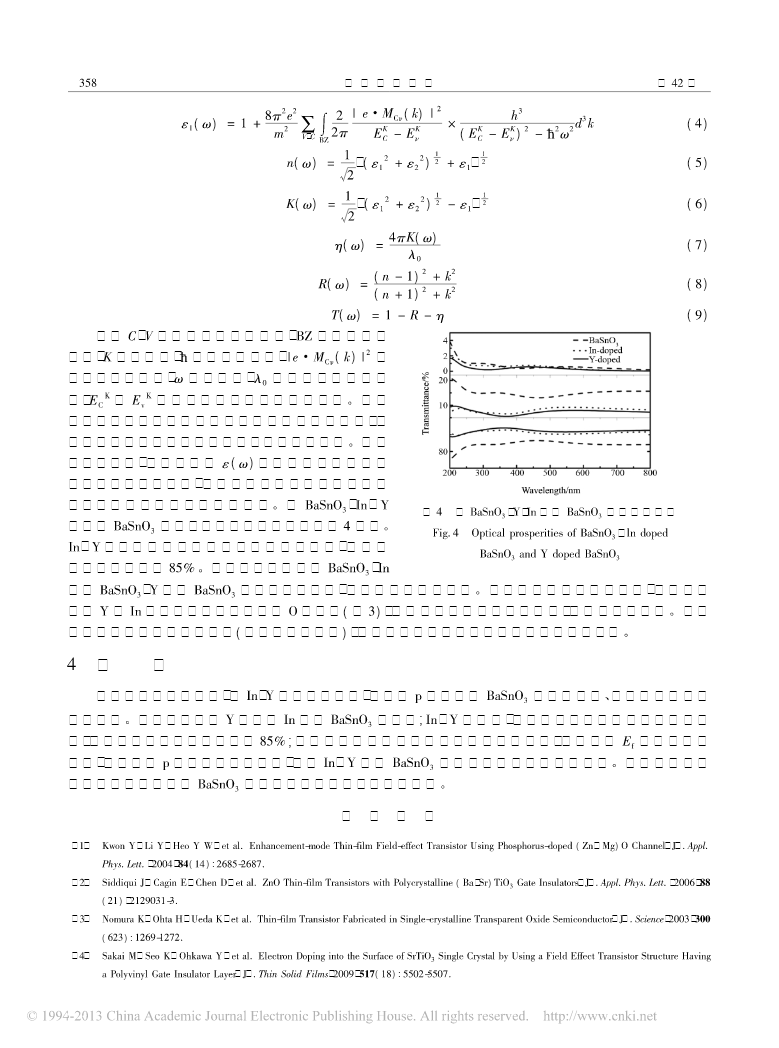
第42卷第2期人工晶体学报Vol.42No.22013年2月JOURNALOFSYNTHETICCRYSTALSFebruary,2013型掺杂的电子结构和光学性能的PBaSnO3第一性原理研究胡诚1,谭兴毅1,2(1.湖北民族学院理学院,恩施445000;2.西北工业大学理学院,凝聚态结构与性质陕西省重点实验室,西安710129)摘要:基于密度泛函理论,计算了以及掺杂的稳定性电子结构和光学性质结果表明以及掺杂YInBaSnO3、。YIn体系结构稳定,且均为型透明导电材料,在可见光区透过率大于,且以及掺杂

P掺杂YVO_4电子结构和弹性性能的第一性原理研究.docx
P掺杂YVO_4电子结构和弹性性能的第一性原理研究随着科技的发展,材料的应用范围也日益扩大,其中,P掺杂YVO_4材料在光催化、传感器和电池等领域有着广泛的应用。为了更好地理解其电子结构和弹性性能,我们采用第一性原理方法进行研究。首先,我们通过密度泛函理论(DFT)的VASP软件包计算了P掺杂后的YVO_4晶体的结构和能带结构。计算结果表明,P掺杂并不能对晶体结构造成明显的影响,但能带结构中出现了新的能级,表明P掺杂引入了新的电子态。接着,我们计算了P掺杂YVO_4的电子密度图和态密度图。通过这些计算结果

(Co,P)双掺杂MgF_2电子结构和光学性质的第一性原理研究.docx
(Co,P)双掺杂MgF_2电子结构和光学性质的第一性原理研究现代材料科学中,双掺杂技术是制备高性能材料的重要手段之一。研究双掺杂对材料的电子结构和光学性质的影响,对于深入了解材料本质和改善材料性能有着重要意义。本文以(Co,P)双掺杂MgF_2电子结构和光学性质的第一性原理研究为题目,综合阐述(Co,P)双掺杂对MgF_2材料的影响。一、研究背景多种金属离子和X向MgF_2中另一原子的双掺杂已被证明对MgF_2材料的性能改善具有显著作用。例如,Li、Na和K离子的单掺杂可以显著提高MgF_2的抗辐射性能

GaSb中p型缺陷电子结构和光学性质的第一性原理研究.docx
GaSb中p型缺陷电子结构和光学性质的第一性原理研究GaSb是一种重要的半导体材料,由于其具有优异的电学和光学性能,被广泛应用于高速电子学、激光器、太阳能电池等领域。其中,p型缺陷是影响器件性能的主要因素之一。因此,对于GaSb中p型缺陷的电子结构和光学性质的研究具有重要的理论价值和应用价值。本文利用第一性原理计算方法,对于GaSb中p型缺陷的电子结构和光学性质进行了详细研究。首先,我们通过VASP软件包在PBE泛函下对GaSb的晶格参数进行了优化。计算结果表明,优化后的晶格参数与实验值较为接近,证明了计